男孩,9岁,骨髓增生异常综合征(MDS)病史一年半后转为急性髓系白血病,同时合并严重肺部感染及噬血细胞综合征。收入北京博仁医院(以下简称我院)后给予噬血治疗的同时积极抗感染,在噬血控制、肺部病灶不再进展时及时行挽救性半相合造血干细胞移植。目前移植后八个月,原发病持续完全缓解,肺部病灶逐渐吸收过程中,造血重建后无合并症发生。
病例简介
男孩,9岁,骨髓增生异常综合征(MDS)病史一年半后转为急性髓系白血病,同时合并严重肺部感染及噬血细胞综合征。收入北京博仁医院(以下简称我院)后给予噬血治疗的同时积极抗感染,在噬血控制、肺部病灶不再进展时及时行挽救性半相合造血干细胞移植。目前移植后八个月,原发病持续完全缓解,肺部病灶逐渐吸收过程中,造血重建后无合并症发生。
患儿:李XX,男,9岁,2017年8月因“面色苍白”起病,外院查血常规:WBC 2.93×10^9/L↓、 Neu 0.97×10^9/L↓、Hb 69g/L↓、PLT 37×10^9/L↓。
外院骨穿MICM诊断为MDS(RAEB-T),染色体:45,X,-Y,[18]/46,XY[2]。FISH:5号染色体相关EGR1基因缺失阳性,提示5q-。
先后就诊于多家医院,予抗感染、输血、口服中药(具体不详)等支持治疗近1年2个月。2018年9月起患儿持续发热、三系减低更为显著。
2018年10月25日因持续高热1个月,先后给予多种抗细菌治疗无效转入我院。入我院后诊治经过:
入院时血常规:WBC 1.89×10^9/L↓、Neu 0.90×10^9/L↓、Hb 40g/L↓、PLT 23×10^9/L↓。肝肾功能未见异常,LDH 937 U/L↑,铁蛋白132.5ng/ml。G试验105.8pg/ml;GM试验阴性;PCT0.29ng/ml。血细、真菌培养阴性;血浆CMV、EBV阴性。
骨髓MICM(2018-10-31):骨髓增生减低,原始髓系细胞占30.5%,外周血原始髓系细胞占22%,考虑MDS转急性髓系白血病;组化:PAS 54%阳性(弥散-细颗粒状),NBE 13%阳性(+),NaF 54%抑制,CE 阴性(-),MPO 11%阳性。
流式:可见26.31% AML细胞,表达CD34,CD13,CD33,HLA-DR,CD123,CD7;部分表达CD117,CD56,不表达CD16,CD19,CD11b,CD2,CD15,CD4,CD14,CD10和CD64。
融合基因:白血病43种常见融合基因筛查阴性。
染色体:45,X,-Y,[1]/45,idem,der(6),t(1;6)(p21;q21)[19]
诊断:1、MDS转急性髓系白血病;2、肺部感染。
考虑到患儿MDS转化为急性髓系白血病,肺部感染严重,三系极度低下,化疗有可能导致感染的进一步加重,且MDS转化的急性髓系白血病化疗效果差,故未予化疗,给予积极抗感染治疗。
先后给予美平、利奈唑胺抗细菌联合伏立康唑、两性霉素B抗真菌治疗后,患者体温未见明显下降且复查CT感染病灶逐渐增大(见图1),后请感染影像专家会诊考虑为肺炎型肺结核,给予四联抗结核治疗后体温逐渐降至正常。
抗结核治疗过程中血象三系逐渐恢复(见图2)。
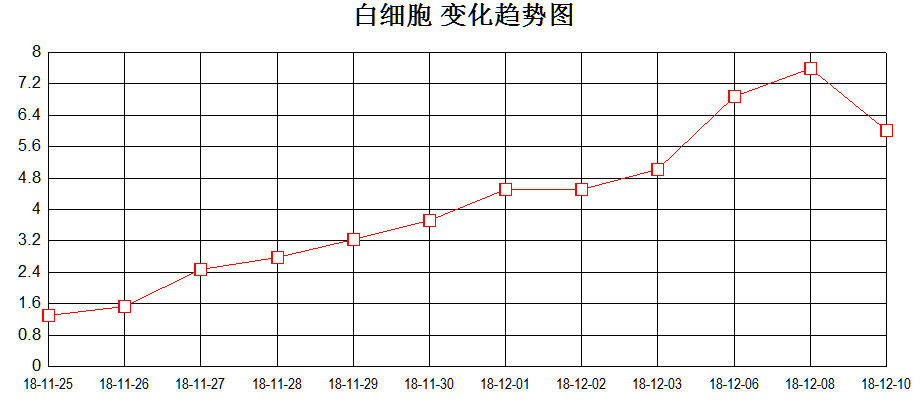
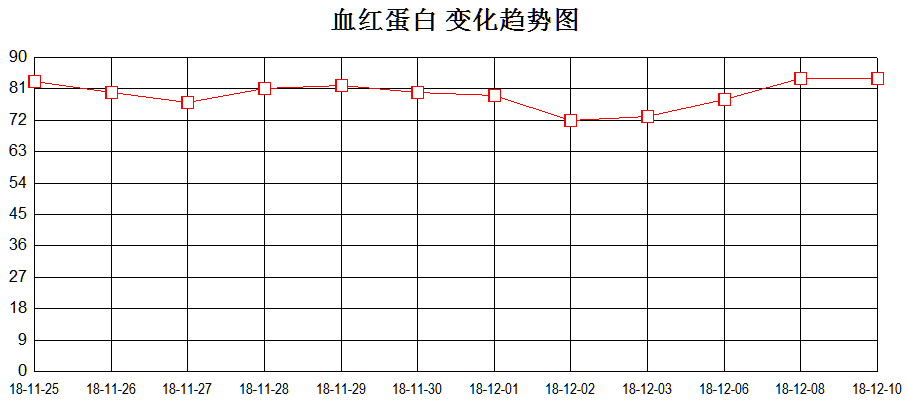
 图2、血象三系变化趋势图
图2、血象三系变化趋势图
CRP、PCT、G试验呈下降趋势。LDH降至正常范围。2018年12月4日复查胸部CT可见感染病灶明显好转(见图3)。
12月25日起患儿再次持续高热,伴肝脾明显肿大。血常规:WBC 0.94×10^9/L↓、Neu 0.45×10^9/L↓、Hb 93g/L↓、PLT 40×10^9/L↓;肝酶升高:ALT 986U/L ↑,AST 856U/L↑ ,纤维蛋白原0.4g/L↓,血清铁蛋白41024.4ng/mL↑,sCD25 4076 pg/ml↑。综合患者血象三系明显下降,转氨酶(见图4)、铁蛋白、sCD25进行性升高,凝血功能异常,肝脾大,且患者携带UNC13D杂合突变,考虑噬血细胞综合征爆发。
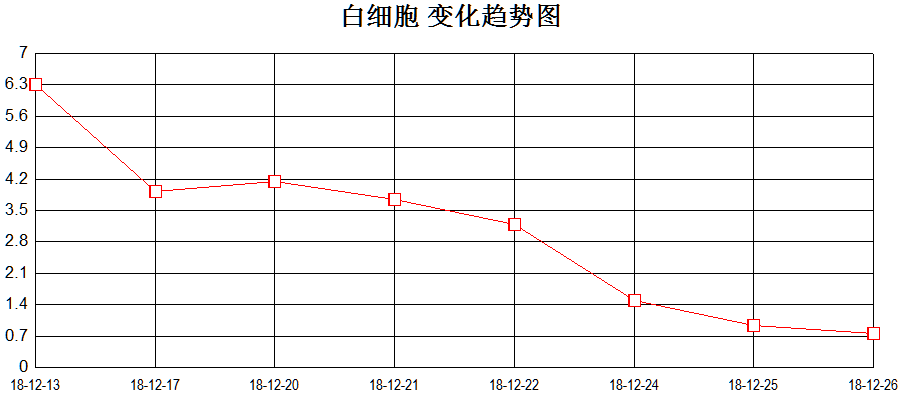
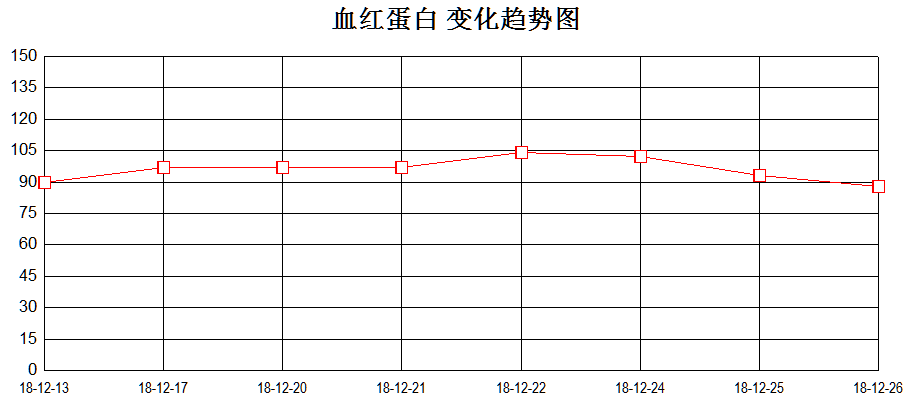
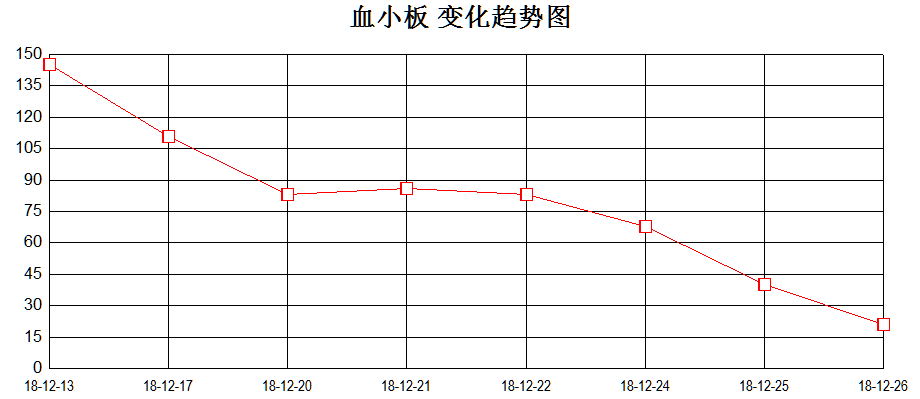
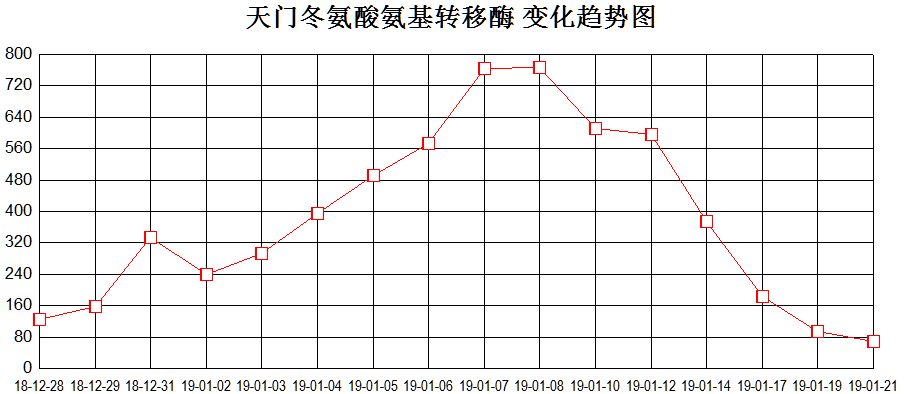
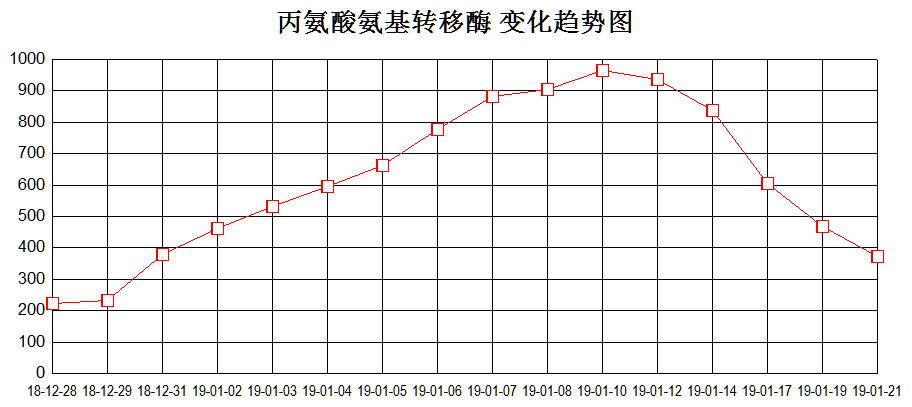
图4、血象三系、转氨酶变化趋势图
先后予激素、MTX、VP16、芦可替尼控制噬血后体温逐渐正常,但血常规未完全恢复,多次复查外周血原始细胞20-30%,骨髓形态原始粒细胞占37%;流式40.21%为异常表型髓系原始细胞。考虑到患儿肿瘤负荷较高且肺部感染没有完全控制、三系减低,MDS转化为AML化疗效果不佳且有可能骨髓抑制期导致感染进一步加重,故与患儿家属充分沟通并告知移植获益与风险后选择异基因造血干细胞移植。患儿没有同胞全合供者,骨髓库没有相合的非血缘供者,结合家族史、遗传易感基因及造血和免疫功能检查等结果最终选定父亲为供者。
患儿于2019年2月13日开始行父供子,HLA 5/10相合,血型A+供A+的挽救性半相合造血干细胞移植。预处理方案为VP-16+Ara-C+BU+FLU+ATG,+15天血小板植活,+16天白细胞植活。移植后1个月复查骨髓及脑脊液残留均为0;骨髓及外周血嵌合率均为完全供者型。造血重建后无合并症发生,现移植后八月余,病情稳定,胸部CT显示感染灶持续好转中(见图5),本病持续完全缓解。
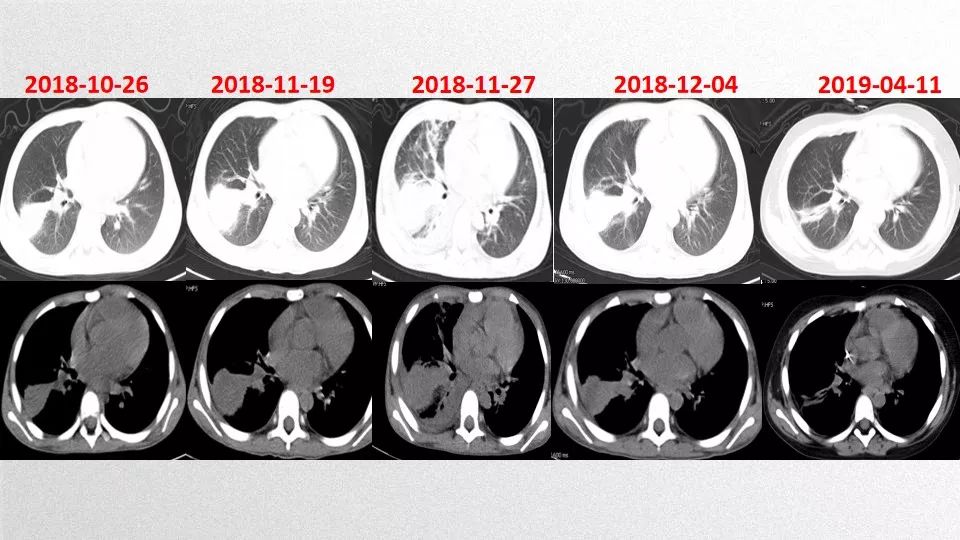
图5、移植后胸部CT显示感染灶持续好转
如何通过解读患者遗传易感基因报告从而分析病情,指导治疗,判断预后及选择供者?
我院应用NGS WES 测序分析700+种血液和免疫系统疾病相关遗传易感基因,基因测序深度> 150x。结果提示,患儿具有明确意义的基因变异有三种:PTPN11,TET2,UNC13D。
1、PTPN11,突变位点p.G503V位于PTP Domain,突变频率84.96%(见图6),经一代测序验证为杂合突变(见图7),而患儿父母都为野生型,说明此变异为体细胞突变,是肿瘤来源的。
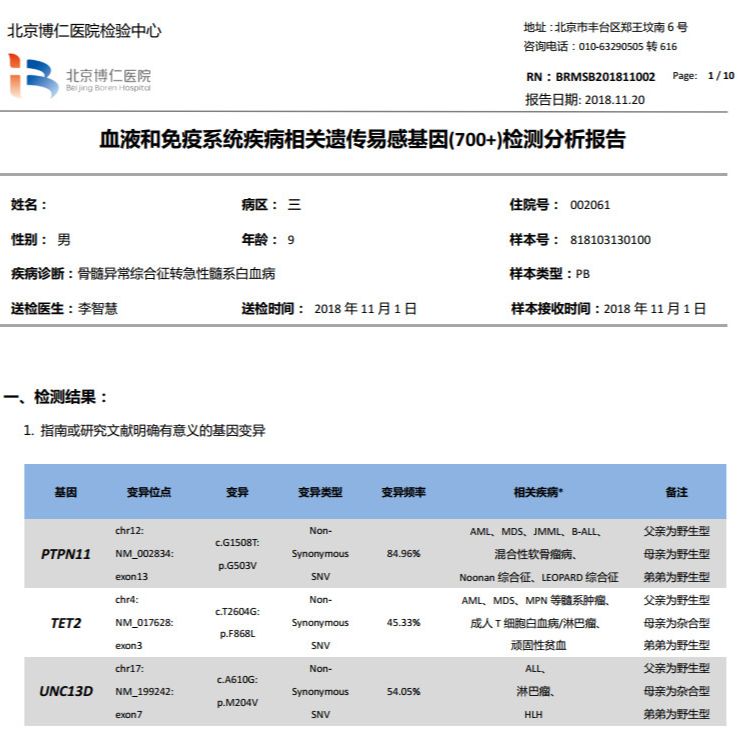
图6、PTPN11突变位点测序
2、TET2突变位点位于p.F868L,变异频率45.33%,其母是杂合型,考虑为胚系来源突变。图8-1示:TET2基因突变在各种血液肿瘤中发生的频率,其中在AML和MDS中的发生频率为23~25%。图8-2示:当携带TET2突变的干细胞自我克隆增殖时,不会引起癌变,如果其克隆扩增的过程中遇到附加突变,则会导致肿瘤的发生。图8-3示:TET2具有免疫调节功能,可以调节免疫细胞分化。当TET2突变功能丧失,就会导致T细胞分化受阻,Th1、Th17细胞增多,Treg细胞减少,导致INF-γ、IL-10分泌下降,IL-17分泌增多;TET2突变功能缺失导致不成熟B细胞凋亡受抑,浆细胞生成增多,抑制免疫球蛋白IgG、Igγ分泌,故患者容易感染;TET2可以抑制巨噬细胞功能,当其突变功能缺失会导致巨噬细胞活化产生更多炎症因子如IL-1b,IL-6,Arg1。
以上对TET2基因功能的解读从一定程度上解释了患儿为什么易发感染及细胞因子风暴。
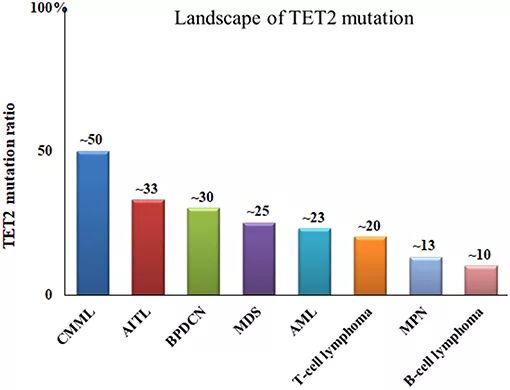 图8-1、TET2基因突变在各种血液肿瘤中发生情况
图8-1、TET2基因突变在各种血液肿瘤中发生情况
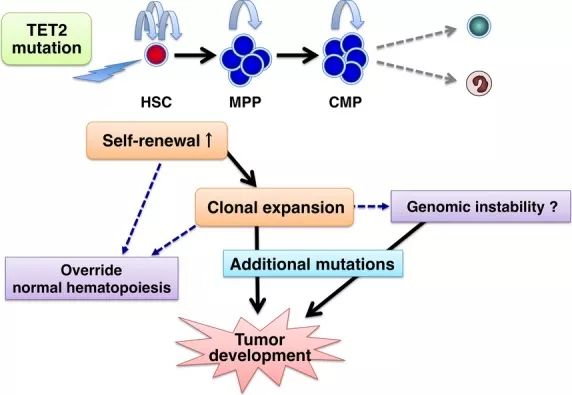
3、UNC13D基因是明确的原发性噬血细胞综合征相关基因,突变位点位于p.M204V,突变频率54.05%,其母亲为杂合型,说明为胚系来源突变。如图9所示,UNC13D是与CD107a脱颗粒功能密切相关的基因,CD107a是囊泡膜蛋白的主要成分,当UNC13D突变并不影响分泌性颗粒的极化以及囊泡与靶细胞膜的锚定,但损伤了启动囊泡及接下来的溶细胞性酶的释放,导致靶细胞无法被正常杀灭。靶细胞分泌抗原使得各种免疫细胞持续活化,不断分泌细胞因子和趋化因子,如干扰素? (IFN-?)、肿瘤坏死因子ɑ(TNF-ɑ)、IL-6、IL-8、IL-10、IL-12、IL-18和巨噬细胞集落刺激因子等,产生严重的“炎症因子风暴”。此外,除了细胞毒性,UNC13D突变还影响血小板颗粒胞吐及中性粒细胞的吞噬体成熟,从而导致凝血及细胞内杀菌作用受损。
对于UNC13D基因的解读更让我们理解了该患儿虽诊断AML,病程中为何容易诱发噬血细胞综合征的原因。
图9、UNC13D突变影响囊泡启动及溶细胞性酶的释放,导致靶细胞无法被正常杀灭
通过这例患儿的诊治经过,我们总结经验如下:
1、MDS称为白血病前期,转变为白血病的可能性很大。由MDS转化的急性白血病患者对常规化疗多不敏感,不容易取得完全缓解,且化疗难以治愈。而这类患者化疗后骨髓抑制期较长,容易发生感染,治疗常非常棘手。因此,这类患者在一般状况允许时,应积极创造条件及时接受异基因造血干细胞移植,这是目前治愈本病的唯一方法。
2、噬血细胞综合征是一种由遗传性或获得性免疫缺陷导致的异常过度免疫反应所引起的一系列临床表现及化验异常,包括发热、全血细胞减少、肝功能异常、肝脾肿大、凝血功能异常、细胞因子及铁蛋白升高等。根据触发因素不同,分为“原发性/遗传性”和“继发性/获得性”两大类。治疗策略分为两个主要方面:短期策略以控制过度炎症状态为主,长期策略以纠正潜在的免疫缺陷为主。对于具有原发性噬血细胞综合征相关基因且反复发作噬血的患者应通过异基因造血干细胞移植重建正常的免疫系统以达到治愈的目的。
3、对于血液病患者,粒缺期长且合并肺部严重感染,抗细菌、真菌治疗无效时,要考虑相对少见的病原菌感染,积极行影像学、病原学等多方面检查,及时更换抗感染药物。
参考文献
[1] Henter JI, Elinder G, Soder O, et al. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood. 1991;78: 2918-2922.
[2] Ishii E, Ohga S, Imashuku S, et al.Review of hemophagocytic lymphohistiocytosis (HLH) in children with focus on Japanese experiences.Crit Rev Oncol Hematol. 2005;53(3):209-223.
[3] Janka, GE, Lehmberg K. Hemophagocytic syndromes - An update. Blood Rev. 2014; 28(4):135-142.
[4] Nakamura L, Bertling A, Brodde MF, et al. First characterization of platelet secretion defect in patients with familial hemophagocytic lymphohistiocytosis type 3 (FHL-3). Blood. 2015; 125(2):412-414.
副主任医师
高博医疗集团 北京博仁医院
毕业于首都医科大学,医学博士,有10余年的血液内科临床工作及实验室研究经验,曾先后在北京友谊医院,北京京都儿童医院等单位就职,擅长白血病及噬血细胞综合征的诊断和治疗及异基因造血干细胞移植,对移植及其合并症的处理具有较丰富的经验。先后在国内外学术期刊发表多篇学术论文。
主任医师,北京博仁医院医疗院长,血液二科(移植技术)主任
中华医学会血液学分会会员
美国血液学会会员
国际血液学会会员
亚太骨髓移植学会(APBMT)会员及学术委员会委员
中国抗癌协会血液肿瘤专业委员会副主任委员(前任)及移植与细胞治疗学组副组长
女医师协会临床肿瘤专业委员会青年委员及血液淋巴肿瘤专业委员会常委
中国医师协会整合医学分会整合血液病专业委员会委员
《中华血液学杂志》编委
《Hematology/Oncology and Stem Cell Therapy》编委
有30余年血液内科,尤其是造血干细胞移植临床工作经验,曾在美国国立卫生研究院心肺血液所做造血干细胞基因治疗领域的博士后研究4年
?






 京公网安备 11010502033352号
京公网安备 11010502033352号